The time series of a chemical concentration necessarily depends on its initial conditions; i.e. the concentration at time 0. An analogous statement is true for gating variables, etc. How do we specify this?
Option 1: NEURON and NMODL defaults
If the species corresponds to one with initial conditions specified by NMODL (or in the case of sodium, potassium, or calcium with meaningful NEURON defaults), then omitting the initial argument will tell NEURON to use those rules. e.g.
from neuron import h, rxd
soma = h.Section(name='soma')
cyt = rxd.Region(h.allsec(), name='cyt', nrn_region='i')
ca = rxd.Species(cyt, name='ca', charge=2, atolscale=1e-6)
na = rxd.Species(cyt, name='na', charge=1)
k = rxd.Species(cyt, name='k', charge=1)
unknown = rxd.Species(cyt, name='unknown', charge=-1)
h.finitialize(-65)
print('ca: %g mM' % ca.nodes[0].concentration)
print('na: %g mM' % na.nodes[0].concentration)
print('k: %g mM' % k.nodes[0].concentration)
print('unknown: %g mM' % unknown.nodes[0].concentration)
As shown here, unknown ions/proteins are by default assigned a concentration by NEURON of 1 mM. The atolscale value for calcium has no effect on the initialized value, but is included here as an example of best practice for working with low concentrations.
Importantly, the NEURON/NMODL rules only apply if there is a corresponding classical NEURON state variable. That is, nrn_region must be set and the Species must have a name assigned.
Running what is otherwise the same code without the nrn_region assigned causes everything to default to 0 µM:
from neuron import h, rxd
soma = h.Section(name='soma')
cyt = rxd.Region(h.allsec(), name='cyt')
ca = rxd.Species(cyt, name='ca', charge=2)
na = rxd.Species(cyt, name='na', charge=1)
k = rxd.Species(cyt, name='k', charge=1)
unknown = rxd.Species(cyt, name='unknown', charge=-1)
h.finitialize(-65)
print('ca: %g mM' % ca.nodes[0].concentration)
print('na: %g mM' % na.nodes[0].concentration)
print('k: %g mM' % k.nodes[0].concentration)
print('unknown: %g mM' % unknown.nodes[0].concentration)
For extracellular species, there is no equivalent traditional NEURON state variable (as those only exist within and along the cell), however NEURON's constant initialization parameters for the nrn_region='o' space are used if available; e.g.
from neuron import h, crxd as rxd
# enabled by default in NEURON 7.7+
rxd.options.enable.extracellular = True
ecs = rxd.Extracellular(-100, -100, -100,
100, 100, 100,
dx=20, volume_fraction=0.2, tortuosity=1.6)
# defining calcium on both intra- and extracellular regions
ca = rxd.Species(ecs, name='ca', charge=2)
# global initialization for NEURON extracellular calcium
# in NEURON 7.7+ can just use: h.cao0_ca_ion = 0.42
# but in older versions of NEURON the variable would not have been defined yet
# and is thus unsettable except via a string call to h
h('cao0_ca_ion = 0.42')
h.finitialize(-65)
print('ca: %g mM' % ca.nodes[0].concentration)
We could do something similar using cai0_ca_ion to set the global initial intracellular calcium concentration.
Option 2: Uniform initial concentration
Setting initial= to a Species or State assigns that value every time the system reinitializes. e.g.
from neuron import h, rxd
soma = h.Section(name='soma')
cyt = rxd.Region([soma], name='cyt')
m = rxd.State(cyt, initial=0.47)
h.finitialize(-65)
print('m = %g' % m.nodes[0].value)
Option 3: Initializing to a function of position
The initial= keyword argument also accepts a callable (e.g. a function) that receives a node object. Nodes have certain properties that are useful for assinging based on position, including .segment (intracellular nodes only) and .x3d, .y3d, and .z3d:
Using .segment:
Here we use the morphology c91662.CNG.swc from NeuroMorpho.Org and initialize based on path distance from the soma.
from neuron import h, gui, rxd
h.load_file('stdrun.hoc')
h.load_file('import3d.hoc')
# load the morphology and instantiate at the top level (i.e. not in a class)
cell = h.Import3d_SWC_read()
cell.input('c91662.CNG.swc')
h.Import3d_GUI(cell, 0)
i3d = h.Import3d_GUI(cell, 0)
i3d.instantiate(None) # pass in a class to instantiate inside the class instead
# increase the number of segments
for sec in h.allsec():
sec.nseg = 1 + 2 * int(sec.L / 20)
def my_initial(node):
# set a reference point
h.distance(0, h.soma[0](0.5))
# compute the distance
distance = h.distance(node.segment)
# return a certain function of the distance
return 2 * h.tanh(distance / 1000.)
cyt = rxd.Region(h.allsec(), name='cyt', nrn_region='i')
ca = rxd.Species(cyt, name='ca', charge=2, initial=my_initial)
h.finitialize(-65)

Using position:
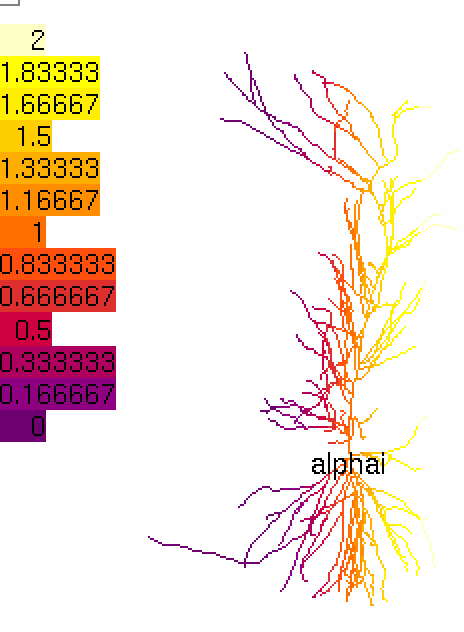
We continue the above example adding a new species, that is initialized based on the x-coordinate. This could happen, for example, on a platform with a nutrient or temperature gradient:
def my_initial2(node):
# return a certain function of the x-coordinate
return 1 + h.tanh(node.x3d / 100.)
alpha = rxd.Parameter(cyt, name='alpha', initial=my_initial2)
h.finitialize(-65)
Option 4: to steady state
Sometimes one might want to initialize a simulation to steady-state where e.g. diffusion, ion channel currents, and chemical reactions all balance each other out. There may be no such possible initial condition due to the interacting parts.
In principle, such initial conditions could be assigned using a variant of the option 3 approach above. In practice, however, it may be simpler to omit the initial= keyword argument, and use an h.FInitializeHandler to loop over locations, setting the values for all states at a given location at the same time. A full example is beyond the scope of this tutorial.